 New
New Haemoglobin Disorders (Thalassaemias and Sickle Cell Disease)
CPDTime.
5m
Published: 08 June 2021


Published: 08 June 2021
Haemoglobin disorders are the most common genetic conditions found in humans, with 7% of the population estimated to be carriers and more than 330 000 infants being born with one of these disorders every year (RACGP 2018; DoH 2019).
People of certain ethnicities are more likely to be carriers of haemoglobin disorders than others. As Australia becomes increasingly multicultural, haemoglobin disorders are expected to become more prevalent (Crighton et al. 2016).
Haemoglobin disorders, also known as haemoglobinopathies, are inherited conditions that affect haemoglobin (Hb) - a protein in the red blood cells responsible for transporting oxygen around the body (TASCA 2020).
These disorders are caused by a mutation in the genes responsible for coding haemoglobin (DoH 2019).
There are over 1000 known gene mutations that can cause haemoglobin disorders, ranging from those that are clinically insignificant to others that have the potential to cause serious illness (GOV.UK 2018).
Haemoglobin disorders include:
(GOV.UK 2018; Crighton et al. 2016)
Among the infants born with a haemoglobin disorder, most inherit sickle cell disease (83%), while about 17% inherit thalassaemias (DoH 2019).
The following groups of people are more likely to be carriers of haemoglobin disorders:
(RACGP 2018)
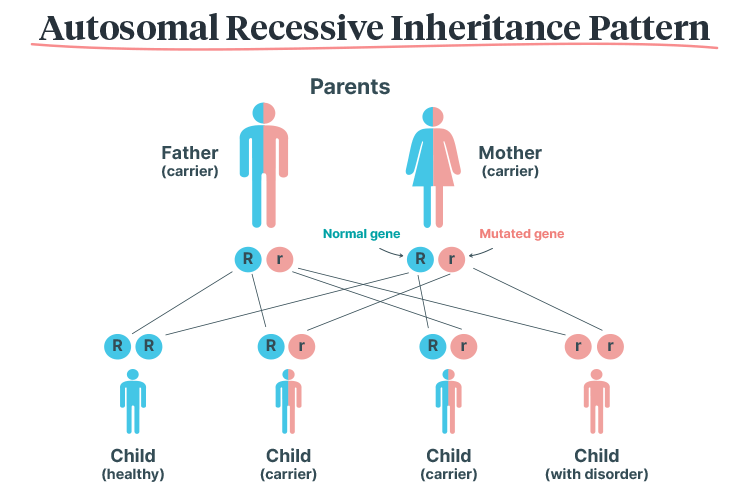
Haemoglobin disorders have an autosomal recessive inheritance pattern, meaning that they can only be passed on if both parents are carrying the mutated gene. The child must inherit two copies of the mutated gene (one from each parent) in order to inherit the disorder. Those who only inherit one copy become carriers (RACGP 2018).
Therefore, if both parents are carriers:
(Mayo Clinic 2020a)
Due to the recessive nature of these conditions, they often occur without any known family history (RACGP 2018).
Haemoglobin disorders can be diagnosed through blood tests and genetic testing (Better Health Channel 2019).
Couples planning a pregnancy can undergo genetic testing to determine whether they are carriers for haemoglobin disorders (RACGP 2018).
The Royal Australian and New Zealand College of Obstetricians and Gynaecologists recommends that all women undergo full blood examination (FBE) for MCV and MCH prior to trying for a pregnancy (or as early as possible in pregnancy). Depending on the results of these tests, as well as other factors such as ethnic background and family history, additional tests may be performed (DoH 2019). These include:
(DoH 2019)
Currently, newborn screening for haemoglobin disorders is not performed in Australia (RACGP 2018).
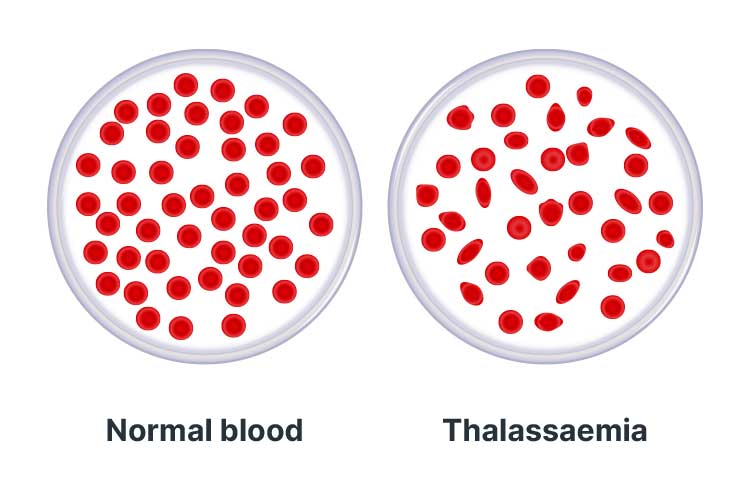
Thalassaemias are a group of haemoglobin disorders that cause insufficient production of haemoglobin, impairing its ability to transport oxygen around the body (GOV.UK 2018).
This means that red blood cells are only able to survive for a few weeks (as opposed to the usual duration of four months), and consequently, cause reduced levels of oxygen in every cell of the individual’s body (Better Health Channel 2019).
There are two types of thalassaemia:
(Better Health Channel 2019)
Thalassaemias are classified as either:
(Better Health Channel 2019)
If left untreated, thalassaemia major can be fatal (Better Health Channel 2019).
(Better Health Channel 2019)
The only known potential cure for thalassaemia major is bone marrow transplant, however, this is associated with serious risks (Better Health Channel 2019).
Despite this, thalassaemia major can be managed through regular blood transfusions alongside medicines to remove excess iron from the blood (Better Health Channel 2019).
Alpha-thalassaemia major, also known as Bart’s Hydrops Fetalis, occurs when no functional alpha-globin genes are inherited (i.e. all four copies of HBA1 and HBA2 are mutated) (GOV.UK 2018).
This is a serious condition that causes anaemia in the fetus and is generally incompatible with life outside of the uterus, leading to stillbirth or death soon after birth. In rare cases, some infants survive through intrauterine transfusions but are likely to develop significant disability (GOV.UK 2018; MedlinePlus 2020).
Bart’s Hydrops Fetalis is also dangerous for the mother and may cause complications during pregnancy, including:
(GOV.UK 2018)
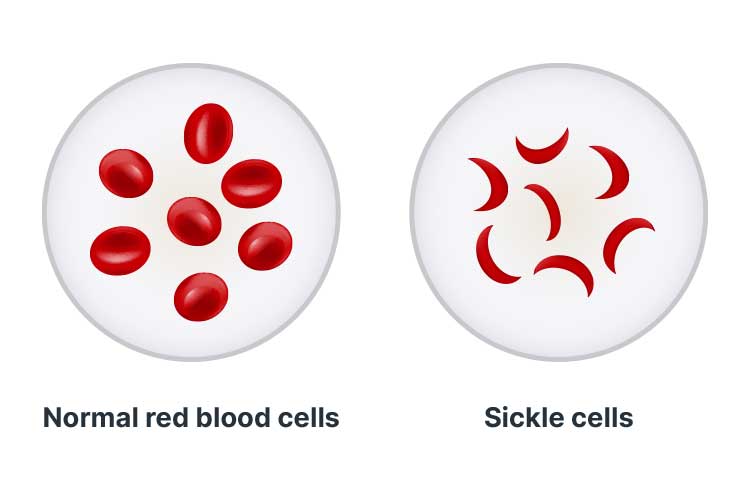
Sickle cell disease (SCD) is an umbrella term describing a group of disorders that cause the production of haemoglobin with an abnormal structure and quality (GOV.UK 2018; CDC 2020).
SCD causes an abnormal process known as sickling, wherein the red blood cells lose oxygen and change shape to resemble a sickle (a farm tool shaped like the letter C) (Lab Tests Online 2017; GOV.UK 2018).
Red blood cells are typically round and can easily manoeuvre through blood vessels. However, cells affected by SCD become hard and sticky, causing them to obstruct blood vessels and block blood flow (Mayo Clinic 2020b).
Normal red blood cells die after 90 to 120 days. However, sickle cells die prematurely (within 10 to 20 days), meaning that there is a constant lack of red blood cells in the body. This causes chronic anaemia (Healthdirect 2020).
SCD is characterised by crises - sudden, potentially life-threatening episodes of pain that usually affect the abdomen, bones or joints. Crises occur when sickle cells obstruct a blood vessel. They may be induced by events such as sudden body temperature changes, dehydration, shortage of oxygen or an infection (Novelli & Gladwin 2016; Healthdirect 2020; GOV.UK 2020).
Other symptoms of SCD include:
(Healthdirect 2020)
Potential complications include:
(Lab Tests Online 2017; GOV.UK 2018; Mayo Clinic 2020b; Healthdirect 2020)
SCD can only be cured through bone marrow or stem cell transplant (GOV.UK 2018).
However, the condition can be managed through:
(GOV.UK 2018; Healthdirect 2020)
Question 1 of 3
Which one of the following conditions is caused by inheriting two abnormal copies of the HBA1 gene and two abnormal copies of the HBA2 gene?


Ausmed Education is a Trusted Information Partner of Healthdirect Australia. Verify here.