 New
New 

Huntington’s disease is a degenerative neurological condition affecting the nerve cells in the brain (Mayo Clinic 2022).
It’s a rare, genetic disease that impairs physical, cognitive and psychological functioning (Mayo Clinic 2022).
The onset of the disease is usually in adulthood, with 90% of people developing symptoms between the ages of 30 and 50. When symptoms appear before the age of 20, it is known as juvenile Huntington’s disease. People who develop the disease early may experience different and faster-progressing symptoms (Mayo Clinic 2022; Yourgenome 2021).
Life expectancy after the onset of symptoms is generally 10 to 30 years, and around 10 for juvenile Huntington’s disease (Mayo Clinic 2022).
What Causes Huntington’s Disease?
Huntington’s disease is caused by a defective HTT gene on chromosome four. The HTT gene is responsible for coding the protein huntingtin. The function of this protein is unclear, but it appears to play a significant role in nerve cell development. Huntingtin is mostly found in the brain but exists in many tissues around the body (MedlinePlus 2020; Yourgenome 2021).
If defective, the HTT gene will create extra repeats of the CAG DNA segment (comprising the cytosine, adenine and guanine bases) in a process known as CAG trinucleotide repeat expansion, causing the huntingtin protein to become abnormally long (MedlinePlus 2020; Yourgenome 2021).
The abnormally long huntingtin protein will separate into smaller, toxic pieces that clump together and gather in the neurons, impairing their normal functioning (MedlinePlus 2020).
People who don’t have Huntington’s disease typically have between 10 and 35 repeats of CAG in the HTT gene. Those with over 36 repeats are at risk of Huntington’s, although some people with 36 to 39 repeats do not end up developing the disease. As a general rule, the more CAG repeats an individual has, the more likely they are to experience earlier onset Huntington’s with more severe symptoms (MedlinePlus 2020; Yourgenome 2021).
Those with over 40 CAG repeats will almost certainly develop Huntington’s, given they have a typical lifespan. While the average number of CAG repeats in someone with Huntington’s disease is about 42, there have been cases of people having up to 180 (Carrol 2016).
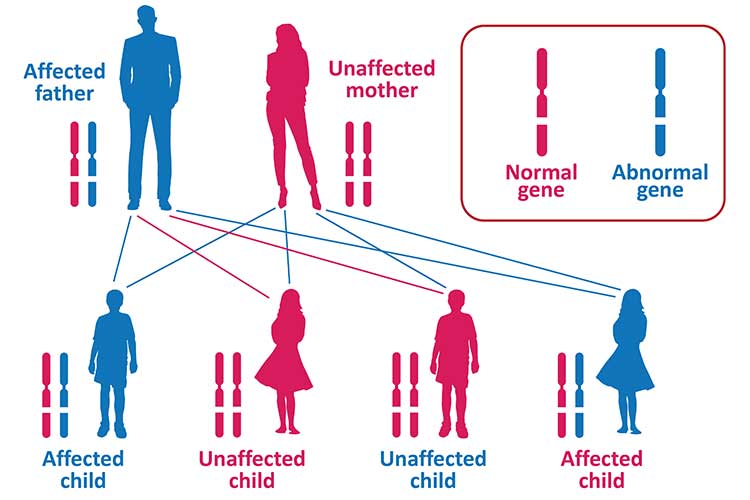
Huntington’s is an autosomal dominant disease, meaning that only one copy of the defective gene is needed for the condition to be inherited. In other words, only one parent needs to have the defective gene in order for their child to potentially inherit it. A parent with the defective gene has a 50% chance of passing it onto their child, and if both parents have the defective gene, this increases to 75% (Mayo Clinic 2022; HDA 2023).
Symptoms of Huntington’s Disease
There are three main types of symptoms: physical, cognitive and emotional. Within these categories, there is a wide range of symptoms that may occur, depending on the individual (Mayo Clinic 2022).
Physical Symptoms
Huntington’s disease can cause issues with both voluntary and involuntary movements. Impairments to voluntary movements, in particular, may significantly affect the individual’s functional capacity (Mayo Clinic 2022). Symptoms may include:
- Chorea (involuntary jerking or writhing)
- Muscle rigidity or dystonia
- Slow or abnormal eye movements
- Issues with gait, posture and balance
- Speech and swallowing difficulties
- Twitching of fingers and toes
- Coordination issues
- Weight loss
- Stiffness
- Fatigue
(Mayo Clinic 2022; Better Health Channel 2022; Healthdirect 2021)
Cognitive Symptoms
- Difficulty organising, prioritising or concentrating on certain tasks
- Preservation (becoming stuck on thoughts, behaviours or actions)
- Decreased control of impulses, potentially leading to outbursts, sexual promiscuity or acting before thinking
- Decreased self-awareness of behaviour and ability
- Difficulty learning new information
- Memory issues
- Decreased speed of thought.
(Mayo Clinic 2022; Better Health Channel 2022; Healthdirect 2021)
Emotional Symptoms
- Depression (which affects about one-third of people with Huntington’s disease)
- Obsessive-compulsive disorder
- Mania
- Bipolar disorder
- Irritability, sadness or apathy
- Mood swings
- Social withdrawal
- Insomnia
- Decreased energy
- Suicidal or death-related thoughts
- Aggression.
(Mayo Clinic 2022; Better Health Channel 2022; Healthdirect 2021)
It’s important to note that depression is associated with changes in brain function caused by Huntington’s disease and is not necessarily a reaction to being diagnosed with the condition (Mayo Clinic 2022).
Juvenile Huntington’s Disease
People with early juvenile Huntington’s disease often present with:
- Concentration difficulties
- A significant and rapid decrease in performance at school
- Behavioural issues
- Muscle rigidity or dystonia
- Tremors of involuntary movements
- Clumsiness and falls
- Seizures.
(Mayo Clinic 2022)

Diagnosis of Huntington’s Disease
Huntington’s disease can be diagnosed using neurological tests, psychological tests, blood tests, genetic tests, and family history (Healthdirect 2021).
Someone with a family history of Huntington’s can elect to undergo a predictive test in order to determine whether they have inherited the defective HTT gene. However, even if the defective gene is present, it is not possible to determine when the symptoms will begin (Mayo Clinic 2022).
Predictive testing can also be performed prenatally through either chorionic villus sampling (CVS) or amniocentesis. However, these tests carry a small risk of miscarriage (UC Davis Health 2016).
Treatment of Huntington’s Disease
Unfortunately, there is no cure for Huntington’s disease and no way to change its course. However, medicines and physical therapy can be used to address certain physical and psychological symptoms (Mayo Clinic 2022; Healthdirect 2021).
Progression of Huntington’s Disease
The rate of disease progression will depend on the individual. In later stages, the individual may require assistance with all aspects of daily life as their nerve cells progressively break down. Towards the end of their life, they will probably be confined to bed. They may be able to understand language and will have some level of awareness of their loved ones, but will most likely be unable to speak (Mayo Clinic 2022).
The most common causes of death are infections such as pneumonia, fall injuries, and complications related to swallowing issues (Mayo Clinic 2022).
Caring for Someone With Huntington’s Disease
The following table contains some suggestions for caring for clients displaying specific symptoms of Huntington’s disease:
| Symptom | Care strategies |
|---|---|
| Loss of motivation |
|
| Difficulty with task sequencing |
|
| Being easily distracted |
|
| Increased carelessness |
|
| Inappropriate social behaviour |
|
| Irritability and aggression |
|
| Communication difficulties |
|
(Better Health Channel 2022)
Test Your Knowledge
Question 1 of 3
Jessie is pregnant with her second child. Her partner and her first child have Huntington’s disease, but she does not. How likely is it that her second child will inherit the disease?
Topics
References
- Better Health Channel 2022, Huntington's Disease, Victoria State Government, viewed 20 November 2023, https://www.betterhealth.vic.gov.au/health/conditionsandtreatments/huntingtons-disease
- Carroll, J 2016, ‘Slightly Long CAG Repeats are More Common Than we Thought’, HDBuzz, 5 July, viewed 20 November 2023, https://en.hdbuzz.net/222
- Healthdirect 2021, Huntington’s Disease, Australian Government, viewed 20 November 2023, https://www.healthdirect.gov.au/huntingtons-disease
- Huntington’s Disease Association 2023, What Causes Huntington's Disease, HDA, viewed 20 November 2023, https://www.hda.org.uk/huntingtons-disease/what-is-huntingtons-disease/genetics-of-huntingtons-disease
- Mayo Clinic 2022, Huntington's Disease, Mayo Clinic, viewed 20 November 2023, https://www.mayoclinic.org/diseases-conditions/huntingtons-disease/symptoms-causes/syc-20356117
- MedlinePlus 2020, Huntington Disease, MedlinePlus, viewed 20 November 2023, https://medlineplus.gov/genetics/condition/huntington-disease/
- UC Davis Health 2016, Genetics - Reproductive Options in HD, UC Davis Health, viewed 20 November 2023, https://health.ucdavis.edu/huntingtons/genetics-prenatal.html
- Yourgenome 2021, What is Huntington's Disease?, Yourgenome, viewed 20 November 2023, https://www.yourgenome.org/facts/what-is-huntingtons-disease